
Coding Cells - Computational tools for Protein Design
Deploying AlphaFold on the Cloud

The coming wave is defined by two core technologies: artificial intelligence (AI) and synthetic biology. Together they will usher in a new dawn for humanity, creating wealth and surplus unlike anything ever seen.
At the center of this wave sits the realization that DNA is information, a biologically evolved encoding and storage system. Over recent decades we have come to understand enough about this information transmission system that we can now intervene to alter its encoding and direct its course. As a result, food, medicine, materials, manufacturing processes, and consumer goods will all be transformed and reimagined. So will humans themselves.
- Mustafa Suleyman, The Coming Wave
CEO @ Microsoft AI, Co-founder DeepMind
Background
This is the first in a series of articles titled ‘Coding Cells’, in which I’ll cover the objectives and technical aspects of my projects in TechBio. In this article, I’ll share my first steps towards building a computational tool for protein design by hosting AlphaFold on a virtual machine (VM) on Google Cloud Platform (GCP).
For the past few months, I’ve been taking the online course “How to Grow Almost Anything” from MIT Media Lab. The course covers both the theory and experimental aspects of synthetic biology and bioengineering, and my favorite topic has been protein design. I learned about the use of AlphaFold, a machine learning model that predicts the 3D structure of a protein for a given sequence of amino acids, and its applications to protein design for biomedical applications.
I was first introduced to computational biology back in undergrad (2018), when I took a course in computational biology that covered algorithmic and statistical techniques for analysis of biomolecules. But back then, AI wasn’t the hot topic like it is today, and applying deep learning to protein structure prediction felt like a theoretical exercise rather than a promising tool for drug discovery.
Today, I’m interested in exploring the use of computational tools and machine learning for protein design in industrial and biomedical applications.
I have been exploring ColabFold, which provides accessible and easy-to-use AlphaFold predictions on Google Colab. Although I’m already familiar with Jupyter notebooks, the ColabFold notebook didn't come without its own set of challenges. The course instructors and TA’s warned us that we should expect the notebook to run for a long time for a large input protein sequence, and to also not be surprised if the notebook times out while running. Running AlphaFold inference for large proteins is computationally expensive, so I was motivated to explore what alternative tools are available for bioengineers and people like me who regularly use AlphaFold through Google Colab.
Google Colab is fine for ad-hoc jobs or initial exploration, but there is an opportunity to build a more comprehensive tool for regular users of AlphaFold, especially protein engineers who use rational design. I decided that a good starting point would be deploying AlphaFold on my own infrastructure on GCP. I plan to expand this project to a suite of tools for the TechBio field, starting with building a beautiful user interface that lets you perform structure predictions, sequence alignments, and visualization all in one software suite.
If you’re having timeout issues with the ColabFold notebook and would prefer to avoid the alternative of deploying ColabFold on your own computer, then this tool will be perfect for you. But if you have a computer with a powerful GPU or would like to host ColabFold on your own cloud infrastructure, then you should stick around for the rest of this article, in which I’ll discuss the steps for deploying ColabFold.
If you’re interested in getting in touch and discussing which direction I should take this project and/or willing to be a beta-tester when the website is live, please fill out this form:
Overview of AlphaFold
Before diving into the technical details of AlphaFold, it’s critical to first understand why protein folding is a such an important concept. If you understand how protein sequence, structure, folding, and function are all related, then you’ll have a greater appreciation of why AlphaFold’s unveiling back in 2021 was so momentous. Check out Levinthal’s Paradox, which I believe is one of the coolest concepts related to biology.
Mustafa Suleyman did a great job of explaining the importance of understanding protein folding.
Proteins are the building blocks of life. Your muscles and blood, hormones and hair, indeed, 75 percent of your dry body weight: all proteins. They are everywhere, coming in every conceivable form, doing myriad vital tasks, from the cords holding your bones together, to the hooks on antibodies used to catch unwanted visitors. Understand proteins, and you’ve taken a giant leap forward in understanding—and mastering—biology.
But there’s a problem. Simply knowing the DNA sequence isn’t enough to know how a protein works. Instead, you need to understand how it folds. Its shape, formed by this knotted folding, is core to its function: collagen in our tendons has a rope-like structure, while enzymes have pockets to hold the molecules they act on. And yet, in advance, there was no means of knowing how this would happen. If you used traditional brute-force computation, which involves systematically trying all the possibilities, it might take longer than the age of the known universe to run through all the possible shapes of a given protein. Finding out how a protein folds was hence an arduous process, holding back the development of everything from drugs to plastic eating enzymes.
-Mustafa Suleyman, The Coming Wave
If you’re interested in learning more about the biology and the emerging TechBio field, check out my article TechBio Adventures, where I discuss in more detail the relationship between protein structure and function.
I won’t cover the details of the model architecture, but if you would like to learn more about these specifics, I suggest checking out the papers I’ve linked in the references section at the bottom of this article. I recommend checking out Google DeepMind’s website discussing their journey to building AlphaFold and how various biotech companies are utilizing AlphaFold for various biomedical applications. For example, researchers are using AlphaFold to understand how protein mutations cause cancer and autism. The one foundational concept I will highlight is that AlphaFold implements the same attention mechanism that you’ll also find at the core of the transformer models that power ChatGPT (OpenAI) and Llama (Meta).
If you’re interested in exploring AlphaFold, here’s the ColabFold notebook that I currently use for generating protein structure predictions.
Google DeepMind recently released AlphaFold 3, the latest iteration of the model which can “generate highly accurate biomolecular structure predictions containing proteins, DNA, RNA, ligands, ions, and also model chemical modifications for proteins and nucleic acids in one platform.”1
Isomorphic Labs was spun-out of DeepMind to entirely focus on applying AI to drug discovery. I’ll be monitoring whether DeepMind open-sources AlphaFold 3 eventually, like it did for the previous 2 iterations. Currently, AlphaFold 3 is closed source and available as a web-service, but only for non-commercial use. This is likely intended to prevent competition with Isomorphic labs’ own drug discovery and development initiatives.
All of this makes for an interesting discussion about the AI value stack, open-source vs proprietary models, and the commoditization of foundation models - I plan on covering these topics in another article. Similar to how Meta open-sourced its line of LLAMA models versus how OpenAI and Anthropic provide programmatic access to their LLMs (large language models), I wonder if we’ll see a big company spend a ton of money to train and open-source a protein structure prediction model, like Meta’s ESMFold. But unlike with LLMs, where there's real revenue to be generated in the enterprise sector, in biotech the value is yet to be realized until someone delivers a viable therapeutic that passes clinical trials where ML models play a significant role in the drug discovery/development process.
Let’s Build
Hardware
Why did I choose Google Cloud Platform?
All my professional experience has been working with Amazon Web Services, so I used this project as an opportunity to get experience with a different cloud platform. All of the tools I used to build the software are open-source, so I didn’t spend much time weighing the advantages and disadvantages of each cloud platform. The benefit of using open-source tools is avoiding vendor lock-in, which is helpful for arbitraging the price of GPUs between different cloud providers.
Virtual Machine specs:
Region: us-west4
Machine type: n1-standard-4
CPU Platform: Intel Skylake
GPU: Nvidia T4
Boot disk: c0-deeplearning-common-gpu-v20240128-debian-11-py310
VM Provisioning model: Standard
Additional disk: 500 GB (Balanced persistent disk)
Check out the GCP documentation to see what regions offer GPU’s
Spot VM’s vs. Standard Instances
At first, I provisioned the VM using the spot provisioning model, which utilizes spare compute resources on GCP In exchange for discounted pricing, the VM can be terminated at any time by GCP. I opted for the spot model at the beginning to save some money, but the VM was terminating so frequently that I eventually had to switch to the standard provisioning model. Spot machines are good for dev work, but once you’re ready for production or can predict your compute needs, a standard machine is better.
Finding GPU resources on GCP
It was tough to find an available GPU on GCP. After trying repeatedly to provision one in the us-central1-f zone, I decided to try again in the early AM, hoping that trying during off-peak hours would improve my luck. After many failed attempts, I was finally able to re-provision my machine by choosing a different region. This time, I set it up in the us-west4 (Las Vegas) region, whereas I had previously set up my spot machine in the us-central1 (Iowa) region. At this stage, latency due to geography isn’t a concern for me, so I can afford to be promiscuous with which GCP regions I choose.
Software
Infrastructure-as-Code
Here are the Terraform resources I used to provision the virtual machine on GCP with the specs listed above:
resource "google_compute_instance" "model_server_instance" {
name = var.machine_name
machine_type = var.machine_type
zone = var.zone
deletion_protection = false
boot_disk {
initialize_params {
image = var.boot_disk_image
size = 100
}
}
attached_disk {
source = google_compute_disk.additional_disk.self_link
device_name = "additional-disk"
}
guest_accelerator {
type = "nvidia-tesla-t4"
count = 1
}
scheduling {
preemptible = false
on_host_maintenance = "TERMINATE"
automatic_restart = true
provisioning_model = "STANDARD"
}
network_interface {
network = "default"
access_config {
nat_ip = data.google_compute_address.existing.address
network_tier = "PREMIUM"
}
}
service_account {
email = var.service_account_email
scopes = ["https://www.googleapis.com/auth/cloud-platform"]
}
metadata = {
service_account_key = google_service_account_key.artifact_registry_reader_key.private_key
enable-serial-console-logging = "true"
}
tags = ["flask-server"]
metadata_startup_script = <<EOF
#!/bin/bash
# Install Docker
echo "Started Model Server Instance..."
sudo apt-get update
sudo apt-get install -y docker.io
# Configure Docker daemon settings
sudo mkdir -p /etc/docker
echo '{
"runtimes": {
"nvidia": {
"path": "nvidia-container-runtime",
"runtimeArgs": []
}
},
"log-driver": "gcplogs",
"log-opts": {
"gcp-meta-name": "ml-model-server-instance"
}
}' | sudo tee /etc/docker/daemon.json
sudo systemctl restart docker
# Authenticate with Google Cloud
gcloud auth configure-docker gcr.io
# Pull the Docker image from Google Artifact Registry
docker pull gcr.io/INSERT_PROJECT_ID_HERE/ml_model_server_image:latest
# Run the Docker image
docker run --gpus all --runtime=nvidia --log-driver=gcplogs -d -p 443:443 --name ml_model_server_container gcr.io/INSERT_PROJECT_ID_HERE/ml_model_server_image:latest
docker exec ml_model_server_container chmod +x /app/install_colabfold.sh
docker exec ml_model_server_container /app/install_colabfold.sh
# Install Nvidia Drivers
sudo /opt/deeplearning/install-driver.sh
EOF
}
# allow rule for port 80
resource "google_compute_firewall" "allow_tcp_80" {
name = "allow-tcp-80"
network = "default"
allow {
protocol = "tcp"
ports = ["80"]
}
source_ranges = ["0.0.0.0/0"]
target_tags = ["flask-server"]
}
# allow rule for port 443
resource "google_compute_firewall" "allow_tcp_443" {
name = "allow-tcp-443"
network = "default"
allow {
protocol = "tcp"
ports = ["443"]
}
source_ranges = ["0.0.0.0/0"]
target_tags = ["flask-server"]
}
resource "google_compute_disk" "additional_disk" {
name = "additional-disk"
type = "pd-balanced"
size = 500
zone = var.zone
image = var.boot_disk_image
}
data "google_compute_address" "existing" {
name = var.reserved_ip_name
project = var.project_id
region = var.region
}
Here’s the Dockerfile for the container that will run our ColabFold model:
FROM nvcr.io/nvidia/tensorflow:24.01-tf2-py3
# Install system dependencies for Colabfold
RUN apt-get update && apt-get install -y \
git \
wget \
curl \
python3-pip
# Set the working directory
WORKDIR /app
COPY install_colabfold.sh /app/
ENV PATH="/app/src/localcolabfold/colabfold-conda/bin:${PATH}"
# Install dependencies for Flask app
COPY requirements.txt /app/
RUN pip3 install --no-cache-dir -r requirements.txt
# Install NVIDIA drivers - uncomment this line if driver installation needed
# RUN sudo /opt/deeplearning/install-driver.shI chose this base image because it came with CUDA, cuBLAS, and cuDNN installed. It’s important to check whether the Nvidia driver version you’re using is compatible with the CUDA version that comes with the container image.
You can find details on what comes with the base image here.
These are the python libraries in the requirements.txt file needed for creating the API layer flask which I will discuss in the next article:
flask
gunicorn
cloud-sql-python-connector[pg8000]
SQLAlchemy
google-cloud-storage
requests
Here’s the Dockerfile that builds on top of the previous Docker image and handles some additional setup (SSL/TLS setup for HTTPS access to the VM, making the installer script executable, starting the flask app which runs the API that gives access to the model).
FROM gcr.io/{INSERT_PROJECT_ID_HERE}/ml_base_image:latest
# Install OpenSSL for certificate generation
RUN apt-get update && apt-get install -y openssl
WORKDIR /app
COPY . /app
# List the contents of the /app directory
RUN ls -la /app
RUN ls -la /app/src
# Make colabfold_batch.sh executable
RUN chmod +x /app/src/colabfold_batch.sh
# Generate self-signed SSL/TLS certificates
RUN openssl req -x509 -newkey rsa:4096 -keyout key.pem -out cert.pem -days 365 -nodes \
-subj "/C=US/ST=State/L=City/O=Organization/CN=example.com"
EXPOSE 443
ENV FLASK_APP=src/app.py
ENV FLASK_RUN_HOST=0.0.0.0
ENV FLASK_RUN_PORT=443
CMD ["gunicorn", "-w", "1", "-b", "0.0.0.0:443", "--certfile=cert.pem", "--keyfile=key.pem", "src.app:app"]Here’s the shell script that clones the repo that I forked from here.
#!/bin/bash
cd src/
wget https://raw.githubusercontent.com/kvaranasi1254/localcolabfold/main/install_colabbatch_linux.sh
bash install_colabbatch_linux.sh
touch COLABFOLD_INSTALLEDCI/CD
The pipeline deploys the code and provisions the infrastructure in the following order:
build_and_push_ml_model_base_image - builds the base container image for the ColabFold model and uploads image to Google Artifact Registry
build_and_push_ml_model_server - builds the container image that builds on top of the base image and uploads the image to Google Artifact Registry
terraform - runs terraform init, plan, and apply to provision resources in GCP
deploy_ml_model_server - connect to VM using SSH and runs commands in the shell to start the container and execute the model installer script
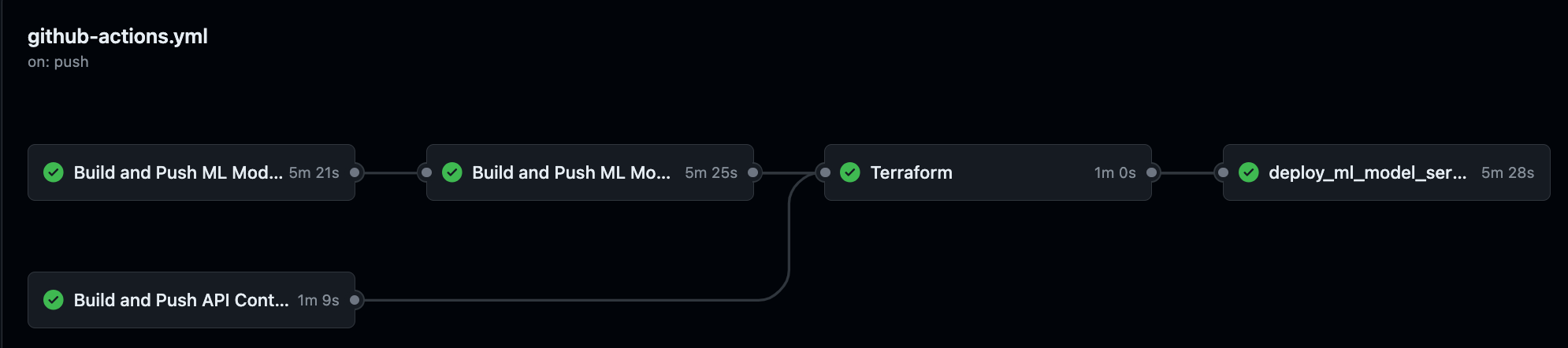
The pipeline executes when code is pushed to the main branch or a pull request is merged into the main branch. If I were on a team of software developers, I would set it up to prohibit pushes to the main branch directly, so that I could enforce peer review of code changes.
I also included the option to only run the terraform steps so that changes that just provisioned new infrastructure would avoid unnecessarily building and deploying the container images.
Here’s the Github actions file to setup the deployment pipeline:
name: Deploy TechBio Project
on:
push:
branches:
- main
pull_request:
branches:
- main
workflow_dispatch:
inputs:
tf-only:
description: "Run Terraform only"
required: false
default: false
type: boolean
build_and_push_ml_model_base_image:
if: ${{ github.event.inputs.tf-only == false}}
name: "Build and Push ML Model Base Container Image"
environment: production
runs-on: ubuntu-latest
steps:
- name: Checkout code
uses: actions/checkout@v2
- name: Remove unnecessary files
run: |
sudo rm -rf /usr/share/dotnet
sudo rm -rf "$AGENT_TOOLSDIRECTORY"/
- name: Set up Cloud SDK
uses: google-github-actions/setup-gcloud@v0.2.0
with:
project_id: ${{ env.PROJECT_ID }}
service_account_key: ${{ secrets.GOOGLE_CREDENTIALS }}
export_default_credentials: true
- name: Configure Docker to use the gcloud command-line tool as a credential helper
run: gcloud auth configure-docker
- name: List ml-model directory contents
run: ls -l ./ml-model/
- name: Build the base Docker image
run: docker build -f ./ml-model/Dockerfile.base -t gcr.io/${{ env.PROJECT_ID }}/ml_base_image:latest ./ml-model/
env:
PROJECT_ID: INSERT_PROJECT_ID_HERE
- name: Push the base Docker image to Google Container Registry
run: docker push gcr.io/${{ env.PROJECT_ID }}/ml_base_image:latest
env:
PROJECT_ID: INSERT_PROJECT_ID_HERE
build_and_push_ml_model_server:
if: ${{ github.event.inputs.tf-only == false}}
name: "Build and Push ML Model Server Container Image"
needs: [build_and_push_ml_model_base_image]
environment: production
runs-on: ubuntu-latest
steps:
- name: Checkout code
uses: actions/checkout@v2
- name: Remove unnecessary files
run: |
sudo rm -rf /usr/share/dotnet
sudo rm -rf "$AGENT_TOOLSDIRECTORY"/
sudo rm -rf /opt/ghc
sudo rm -rf "/usr/local/share/boost"
sudo rm -rf /opt/hostedtoolcache
- name: Set up Cloud SDK
uses: google-github-actions/setup-gcloud@v0.2.0
with:
project_id: ${{ env.PROJECT_ID }}
service_account_key: ${{ secrets.GOOGLE_CREDENTIALS }}
export_default_credentials: true
- name: Configure Docker to use the gcloud command-line tool as a credential helper
run: gcloud auth configure-docker
- name: Build the Docker image
run: docker build --tag gcr.io/${{ env.PROJECT_ID }}/ml_model_server_image:latest ./ml-model/
env:
PROJECT_ID: INSERT_PROJECT_ID_HERE
- name: Push the Docker image to Google Container Registry
run: docker push gcr.io/${{ env.PROJECT_ID }}/ml_model_server_image:latest
env:
PROJECT_ID: INSERT_PROJECT_ID_HERE
terraform:
name: "Terraform"
needs: [build_and_push_ml_model_server, build_and_push_api]
if: always()
runs-on: ubuntu-latest
environment: production
# Use the Bash shell regardless whether the GitHub Actions runner is ubuntu-latest, macos-latest, or windows-latest
defaults:
run:
shell: bash
steps:
# Checkout the repository to the GitHub Actions runner
- name: Checkout
uses: actions/checkout@v3
# Install the latest version of Terraform CLI and configure the Terraform CLI configuration file with a Terraform Cloud user API token
- name: Setup Terraform
uses: hashicorp/setup-terraform@v1
with:
cli_config_credentials_token: ${{ secrets.TF_API_TOKEN }}
# Initialize a new or existing Terraform working directory by creating initial files, loading any remote state, downloading modules, etc.
- name: Terraform Init
run: terraform init
# Generates an execution plan for Terraform
- name: Terraform Plan
run: terraform plan -input=false
# Apply the changes required to reach the desired state of the configuration
- name: Terraform Apply
run: terraform apply -auto-approve -input=false
deploy_ml_model_server:
needs: [build_and_push_ml_model_server, terraform]
runs-on: ubuntu-latest
timeout-minutes: 60 # Adjust this value based on the needs
steps:
- name: SSH and Deploy
uses: appleboy/ssh-action@master
with:
host: ${{ secrets.VM_IP }}
username: ${{ secrets.VM_USERNAME }}
passphrase: ${{ secrets.VM_SSH_KEY_PASSPHRASE }}
key: ${{ secrets.VM_SSH_KEY }}
script: |
set -e # Exit immediately if a command exits with a non-zero status.
docker pull gcr.io/${{ env.PROJECT_ID }}/ml_model_server_image:latest
docker stop ml_model_server_container || true
docker rm ml_model_server_container || true
docker run --gpus all --runtime=nvidia --log-driver=gcplogs -d -p 443:443 --name ml_model_server_container gcr.io/${{ env.PROJECT_ID }}/ml_model_server_image:latest
docker exec ml_model_server_container chmod +x /app/install_colabfold.sh
docker exec ml_model_server_container /app/install_colabfold.sh
# Wait for a few seconds to ensure Docker has enough time to start the container.
sleep 5
# Check if the container is running.
docker container inspect ml_model_server_container -f '{{.State.Running}}' | grep true
env:
PROJECT_ID: INSERT_PROJECT_ID_HERETesting
Here, we SSH’d into the VM and ran the ‘docker ps’ command to see if the container is up and running:

You can use ‘docker logs CONTAINER_ID’ to see the logs for the container if you’re having trouble running it.
Before setting up the API, I tested the ColabFold model by . Run ‘docker exec -it ml_model_server_container bash’ to enter the shell inside the container
Here’s the terminal output after running ColabFold for Human Myoglobin (P02144), a protein found in cardiac and skeletal tissue in almost all mammals. I’ll direct you towards the ColabFold paper for a detailed explanation on the model outputs, the purpose of the recycles, and how to use pLDDT as a measure of the the confidence of the model’s predictions.
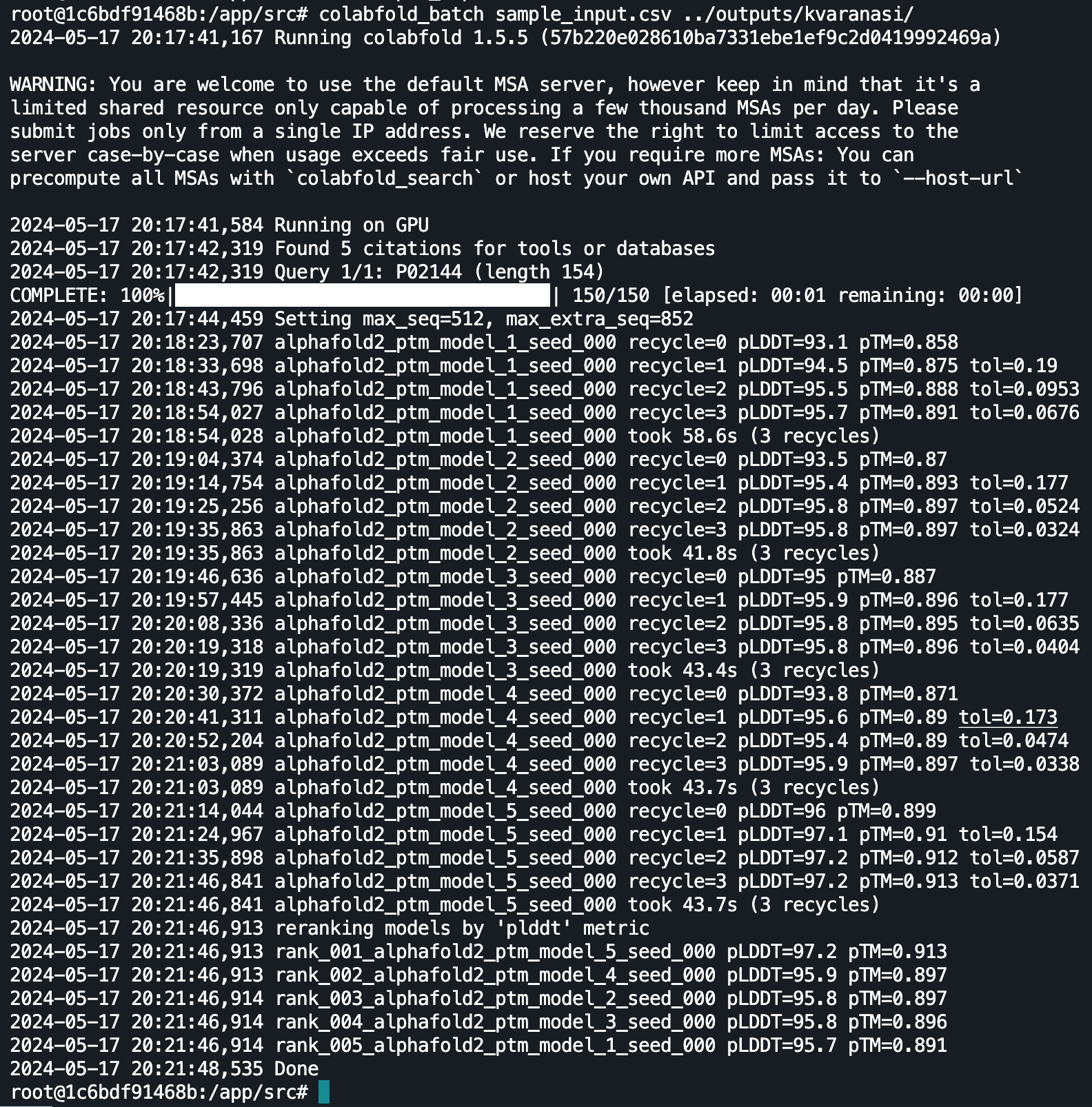
The key output for us is the pdb (standard file format for atomic coordinates) file, which we can for further exploration in tools like PyMol, but for the purposes of this article I opted for the RCSB tool.

Looking Ahead
The next step for the project will be to build an API that protein engineers can use to run AlphaFold jobs. After discussing with a few people, I learned that having sequence alignments, molecular visualization, and structure predictions all in one interface would be useful. The API layer will also be needed for a frontend web application that offers all these features. I’m thinking of even adding protein-docking features eventually, and in the long-term it would be interesting to integrate every open-source model out there and provide value at the application layer by building a managed service for bioengineers and synthetic biologists to use.
Thanks for reading, and leave a comment if you have any feedback. Feel free to share this article with whomever you think would be interested.
Special thanks to Sravya Varanasi for proofreading drafts of this article.
Special thanks to Likhit Dharmapuri for guidance on the app architecture.
Resources
Papers
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024). https://doi.org/10.1038/s41586-024-07487-w
Mirdita, M., Schütze, K., Moriwaki, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682 (2022). https://doi.org/10.1038/s41592-022-01488-1
https://golgi.sandbox.google.com/about